Article Text

Abstract
In the 1950s, Bickerstaff and Fisher independently described cases with a unique presentation of ophthalmoplegia and ataxia. The neurological features were typically preceded by an antecedent infection and the majority of patients made a spontaneous recovery. In the cases with Bickerstaff brainstem encephalitis, there was associated altered consciousness and in some, hyperreflexia, in support of a central pathology whereas in Fisher syndrome, patients were areflexic in keeping with a peripheral aetiology. However, both authors recognised certain similarities to Guillain–Barré syndrome such as the presence of peripheral neuropathy and cerebrospinal fluid albuminocytological dissociation. The discovery of immunoglobulin G anti-GQ1b antibodies in patients with Fisher syndrome and later in Bickerstaff brainstem encephalitis was crucial in providing the necessary evidence to conclude that both conditions were in fact part of the same spectrum of disease by virtue of their common clinical and immunological profiles. Following this, other neurological presentations that share anti-GQ1b antibodies emerged in the literature. These include acute ophthalmoparesis and acute ataxic neuropathy, which represent the less extensive spectrum of the disease whereas pharyngeal-cervical-brachial weakness and Fisher syndrome overlap with Guillain–Barré syndrome represent the more extensive end of the spectrum. The conditions can be referred to as the ‘anti-GQ1b antibody syndrome’. In this review, we look back at the historical descriptions and describe how our understanding of Fisher syndrome and Bickerstaff brainstem encephalitis has evolved from their initial descriptions more than half a century ago.
- Bickerstaff brainstem encephalitis
- Fisher syndrome
- anti-GQ1b antibody
- Guillain–Barré syndrome
- neuromuscular
- neuropathy
- ganglioside
- CIDP
- Miller Fisher syndrome
Statistics from Altmetric.com
- Bickerstaff brainstem encephalitis
- Fisher syndrome
- anti-GQ1b antibody
- Guillain–Barré syndrome
- neuromuscular
- neuropathy
- ganglioside
- CIDP
- Miller Fisher syndrome
Introduction
In their article titled ‘Mesencephalitis and rhombencephalitis’ in 1951, Bickerstaff and Cloake described patients who presented with external ophthalmoplegia, ataxia and altered consciousness, preceded by an infective episode, all of whom made good spontaneous recovery.1 In 1957, Bickerstaff went on to expand his case series.2 Prior to the latter description, Miller Fisher in 1956, described his own encounter with patients who had a combination of external ophthalmoplegia, ataxia and areflexia, noting spontaneous recovery.3 There were striking similarities in the observations from both authors in the form of external ophthalmoplegia and ataxia. Although they had both at the time raised the possibility that the underlying immunopathogenesis was similar to Guillain–Barré syndrome (GBS), each syndrome was considered mutually exclusive.
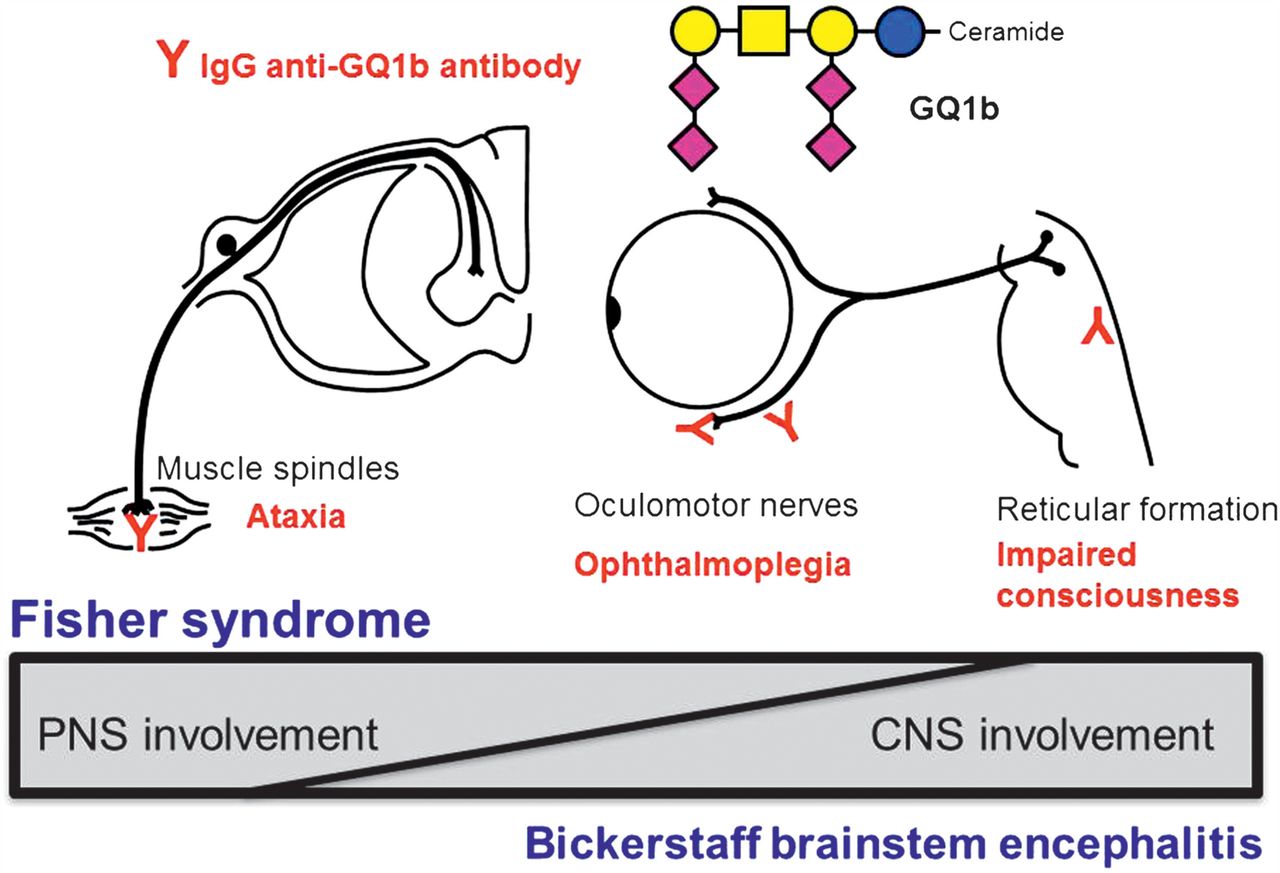
In 1992, the seminal work by Chiba and colleagues in discovering immunoglobulin G (IgG) anti-GQ1b antibodies in Fisher syndrome (FS) patients led to the unravelling of the pathogenesis of FS.4 This was quickly followed by reports of the autoantibodies in patients with Bickerstaff brainstem encephalitis (BBE),5 which brought about the clinical spectrum that is referred to as the ‘Fisher–Bickerstaff syndrome’ (figure 1).6 At the milder end of this clinical spectrum, patients present with only ophthalmoplegia or ataxia but are serologically positive for the IgG anti-GQ1b antibodies. A more inclusive nomenclature such as ‘anti-GQ1b antibody syndrome’ could be used to include the common serological profile7 when referring to the clinical syndromes described by both Bickerstaff and Fisher (table 1). In this review, we look back at the initial descriptions and describe how our understanding of this syndrome has evolved over the last 2 decades.

Fisher–Bickerstaff syndrome. The GQ1b antigen is highly expressed in the oculomotor, trochlear and abducens nerves, muscle spindles in the limbs, and probably reticular formation in the brainstem. Infection by microorganisms bearing the GQ1b epitope may induce production of immunoglobulin G (IgG) anti-GQ1b antibodies in susceptible patients. The binding of anti-GQ1b antibodies to GQ1b antigens expressed on the relevant cranial nerves and muscle spindles induces Fisher syndrome. In some cases, the anti-GQ1b antibodies may also enter the brainstem and bind to GQ1b, inducing Bickerstaff brainstem encephalitis. A continuous spectrum exists between these conditions presenting with variable central and peripheral nervous system (CNS and PNS) involvement. Modified from reference6 with permission.
Anti-GQ1b antibody syndrome
Historical perspective
Original descriptions
The presence of ‘cranial neuropathies of the GBS type’ has been described in the European literature even before the renowned description by Fisher.8 Guillain recognised the various presentations in GBS and at a Belgian symposium in 1938 proposed a clinical classification that takes into account the four topographical presentations: (1) involvement of the extremities only (‘la forme inférieur’); (2) deficits of both extremities and cranial nerves (‘la forme mixte spinale et mésocéphalique’); (3) a syndrome limited to the cranial nerves (‘la forme mésocéphalique pure’); and (4) polyradiculopathy and mentation change (‘une forme de polyradiculonéurite avec troubles mentaux’).9 The third subtype bears some similarities to FS whereas the recognition that mentation can be affected in the fourth subtype is in support of BBE. Guillain described a patient who following an antecedent infection, developed trigeminal, abducens and facial nerve palsies associated with cerebrospinal fluid (CSF) albuminocytological dissociation and subsequent complete recovery. This was followed by similar reports of cases and whereas this clinical entity was widely accepted in Europe, published reports from American and British neurologists were lacking until the 1950s.
In his article in 1956, Fisher described three patients presenting with external ophthalmoplegia and ataxia, commenting on the striking symmetry of the ophthalmoplegia and the severity of the ataxia.3 One patient was also drowsy during the acute phase of the illness. The presence of areflexia and neuropathy along with CSF albuminocytological dissociation were reminiscent of GBS. All three patients made spontaneous recoveries. In support of his theory, he included reports of two patients with GBS but both patients also exhibited signs of ophthalmoplegia (FS overlap with GBS). One patient succumbed to respiratory failure allowing pathological studies to be done. This showed evidence of severe myelin destruction with perivascular mononuclear infiltration and a normal brain stem, features in keeping with an acute infective polyneuritis.
In Bickerstaff's original writings in 1951 and 1957, he highlighted drowsiness as a key denominator in all eight cases.1 ,2 Common to all were also ptosis, ophthalmoplegia (mostly complete) and facial palsy. Other clinical features recorded although not always present include limb ataxia, dysarthria and areflexia (with extensor plantars). One patient also presented with gross flaccid paralysis. CSF analysis showed raised protein although in five patients the cell counts were also raised (more than 10 lymphocytes). Electroencephalography done on two patients revealed slow wave activity indicative of underlying encephalopathic changes. Apart from one patient who died following a convulsive episode, the remaining patients made good recovery. The autopsy findings demonstrated beading and swelling of myelin as well as twinning and swelling of astrocytes, all of which are supportive of an oedematous process. Given the overall good outcome, Bickerstaff postulated an aetiology that resulted in the suppression of function without destroying tissue and rather than a direct infective process, a secondary effect from a systemic illness was more likely. He alluded to the process showing similarities to that of infective polyneuritis.
Central versus peripheral aetiology
In 1982, Bickerstaff as a coauthor in an article by Al-Din et al reported on 18 cases of brainstem encephalitis and FS arguing a central cause for both illness.10 All 18 patients presented with progressive ophthalmoplegia and ataxia. In the majority, the neurological symptoms were preceded by an infective episode. Most patients also had altered consciousness (12 patients) and areflexia (11 patients). The authors suggested the likelihood of a ‘hypersensitivity’ reaction to an infective episode but argued that in FS, the underlying abnormality was in the brainstem based on brain imaging in some that showed hypodensity changes in the brainstem. The ataxia was thought to be cerebellar in origin and areflexia due to mesencephalic and upper pontine reticular formation involvement. BBE was also considered to be clinically distinct from GBS as none of their cases had peripheral involvement including normal neurophysiology but instead there were abnormalities on brain imaging in three patients as well as pathological changes in the brainstem of one patient.
Further arguments for a central cause in FS were made in which the eye movements were described in detail in an FS patient attributing this to a supranuclear cause.11 In the editorial of the same issue, Ropper disagreed with the authors' reasoning, suggesting that a peripheral cause could just as easily, if not more likely, give rise to the observations described.12 The lack of abnormal pathology in the brainstem of GBS cases with ophthalmoplegia and ataxic polyneuropathy were also inconsistent with a central cause. Ropper, in his editorial, went on to criticise the study by Al-Din et al;10 in his opinion, only six of their cases were FS, whereas others were obscure brainstem lesions without peripheral involvement. The authors rebutted Ropper's comments in their letter to the editor,13 stating that the abnormal CT imaging in three patients, abnormal EEG in two and abnormal brainstem evoked potential in one patient were suggestive of a central cause. They also argued that the lack of necropsy findings reflected the benign prognosis seen in FS and BBE.
Following the discovery of the IgG anti-GQ1b antibodies in patients with FS and BBE,4 ,5 it became evident that both conditions were part of a similar spectrum of disease involving an immune-mediated process, triggered by an antecedent infective episode. In a study of 62 patients with BBE, diagnosed by the strict criteria of progressive, relatively symmetrical external ophthalmoplegia and ataxia by 4 weeks, and disturbance of consciousness or hyperreflexia, the authors described positive anti-GQ1b antibody in 66% and abnormal brain MRI in 30% of patients.14 They also noted that in 60% of patients, there was associated flaccid symmetrical tetraparesis. Autopsy findings were available in one BBE patient which showed the presence of definite inflammatory changes in the brainstem with evidence of perivascular lymphocytic infiltration and surrounding oedema in the medulla. Interestingly, neurophysiology showed peripheral motor axonal degeneration. The latter findings were supportive of overlapping GBS in some BBE cases lending further support that a continuous spectrum exists between GBS and BBE.
In a more recent study of a large BBE and FS population (n=53 BBE; 466 FS), the authors demonstrated that FS and BBE were not distinct conditions but share similar clinical and laboratory profile (table 2).15 In the study, impaired consciousness was required for the diagnosis of BBE whereas, clear consciousness and hypo- or areflexia were necessary to make a diagnosis of FS. Although brain MRI and EEG recordings were more likely to be abnormal in BBE patients (11% and 57%, respectively), they were also abnormal in some FS patients (1% and 25%, respectively) despite the fact that the consciousness was retained in FS patients. These observations suggest that central components can occasionally be affected in FS. Peripheral nerve testing demonstrated abnormalities (most frequently the absence of soleus H reflex) in 75% of four BBE and 74% of 28 FS patients. Body sway analyses showed changes indicative of dysfunction within the proprioceptive afferent system in 67% of three BBE and 72% of 18 FS patients. These latter findings would suggest that ataxia in both BBE and FS are caused by a similar mechanism that involves the proprioceptive afferent system rather than cerebellar. Interestingly, 62 patients could not be classified as they did not fulfil the strict criteria of areflexia in FS. However, 58% of this unclassified group were serologically positive for the anti-GQ1b antibodies. The authors proposed a new terminology ‘Fisher–Bickerstaff syndrome’ to include these unclassified conditions as well as FS and BBE. The terminology is helpful in highlighting the clinical continuity among FS, BBE and conditions that may not quite fulfil the full set of criteria. In the clinical setting, however, individual cases could still carry the eponyms FS and BBE.
Clinical and laboratory findings in a cohort of patients with Bickerstaff brainstem encephalitis or Fisher syndrome
Clinical features
Epidemiology
The reported incidence of GBS in Western countries ranges from 0.89 to 1.89 (median, 1.11) cases per 100 000 person-years.16 In comparison with GBS, both BBE and FS are relatively rare. Thus, there are no epidemiology data specifically looking at the prevalence and incidence of FS or BBE. Any available data have been extracted from existing GBS population studies. In the series of GBS cases documented by Ropper et al, FS accounted for 3% of cases.17 In other Western populations, FS is reported to have an incidence of approximately 1%–5% of GBS cases.18 However, the incidence is appreciably higher in Asian countries such as Taiwan (19%) and Japan (25%).19 ,20 There have been no epidemiology studies of BBE with accurate incidence figures to date but our clinical experience suggests that BBE is less frequent.
Common descriptions
The classical triad seen in FS is ataxia, ophthalmoplegia and areflexia. In the event there is associated alteration in the level of consciousness or hyperreflexia, a diagnosis of BBE is preferred as a reflection of the central nervous system involvement (table 1). In clinical practice and also in the original descriptions, there are other clinical symptoms and signs that can also be present in patients with FS and BBE. The most common of these include ptosis, mydriasis, peripheral sensory disturbance and facial palsies (at times presenting as a delayed feature after other features have started to improve).3 ,20
Less extensive forms
There are reports of patients with positive anti-GQ1b antibodies who do not demonstrate the full complement of the FS triad. Instead, these patients have either ophthalmoplegia or ataxia. ‘Acute ophthalmoparesis’ (AO) represents patients presenting with ophthalmoplegia without ataxia or areflexia following an antecedent illness who are seropositive for the anti-GQ1b antibodies.21 ,22 There have also been reports of unilateral or isolated presentations of cranial neuropathies. Ropper in his article, ‘Further regional variants of acute immune polyneuropathy’ included one case of isolated right abducens nerve palsy and paraesthesia associated with abnormal neurophysiology.23 There have since been cases of unilateral ophthalmoplegia and ataxia and unilateral third cranial nerve palsy reported.24 ,25 These cases initially raised the possibility of a vascular pathology but normal imaging, the presence of anti-GQ1b antibodies and spontaneous complete recovery are in keeping with an immune-mediated process. In a study of 100 patients presenting with isolated abducens palsy, 25% of patients were seropositive for IgG anti-GQ1b antibodies.26
In 1962, Richter described an autopsy case of ‘The ataxic form of polyradiculoneuritis (Landry-Guillain-Barré syndrome)’, which is characterised by profound ataxia with negative Romberg sign and no ophthalmoplegia.27 Patients with ataxic GBS carry IgG anti-GQ1b antibodies, suggesting that this condition is an incomplete form of FS.28 ,29 Initially, the nosological position of acute sensory ataxic neuropathy was unclear. However, ataxic GBS and acute sensory ataxic neuropathy were later shown to share similar features: antecedent infectious symptom (86% vs 83%), distal paraesthesia (70% vs 88%), superficial sensory impairment (27% vs 24%), IgG antibodies against GQ1b (65% vs 18%) and GD1b (46% vs 47%) and CSF albuminocytological dissociation (30% vs 39%).30 These suggest that both conditions are part of the same clinical spectrum and could be comprehensively referred to as ‘acute ataxic neuropathy (without ophthalmoplegia)’ to avoid nosological confusion considering that FS is not defined by the absence or presence of sensory ataxia. The presence of areflexia, distal paraesthesia and CSF albuminocytological dissociation provides the link to GBS.
Although ptosis, mydriasis and bulbar palsy are not part of the triad seen in FS, these signs have been reported to occur in both FS and BBE cases.15 Isolated ptosis, mydriasis or oropharyngeal palsy in association with anti-GQ1b antibodies have been reported and these cases are also representative of the less extensive forms of the anti-GQ1b antibody syndrome.31–35
More extensive forms
Ropper described three cases of pharyngeal-cervical-brachial (PCB) weakness, characterised by areflexia and weakness of the oropharyngeal, neck and shoulder muscles.36 This was followed by a further case of PCB overlapped with FS but in this latter case, there was also drowsiness, suggesting that this was more likely to be PCB overlapped by BBE.23 In a retrospective study of 100 PCB patients, 13 patients had pure PCB, 26 patients had FS and five patients had BBE.37 IgG anti-GQ1b antibodies were present in 39% of patients and the majority were FS and BBE overlaps (73% and 60%, respectively). These clinical and serological findings suggest that PCB is in continuity with FS and BBE and can be regarded as a more extensive form of this syndrome.
As previously mentioned, some FS and BBE patients can also develop an overlap of GBS, resulting in significant limb weakness.20 These groups of patients have also been associated with anti-GQ1b antibodies as well as anti-GM1 or -GD1a antibodies and can also be considered to represent the more extensive form of the spectrum.38
Recurrent illnesses
Although FS and BBE are typically monophasic, recurrent episodes have been described in the literature. These are rare and the time between relapses varies. Each episode may be similar in its clinical characteristics as was described in four out of 28 FS patients.39 However, patients can present with different patterns that range in its severity; in two separate case reports, a single patient was noted to develop a BBE pattern of illness (with associated drowsiness and extensor plantars) on the third relapse of her anti-GQ1b antibody syndrome40 whereas in another patient, recovery from the first episode of FS overlapped by GBS was followed by two relapses 2 years apart where the patients exhibited the typical FS phenotype.41
Diagnostic approach
The approach to the diagnosis of FS or BBE lies in the recognition of their unique set of clinical features. The presence of an antecedent illness and distal paraesthesia are often associated with the anti-GQ1b antibody syndrome and there should be a higher index of suspicion for immune-mediated neuropathies when these features are present. However, other possible differential diagnosis may need to be considered which include brainstem stroke, Wernicke encephalopathy, myasthenia gravis and botulism. Careful clinical assessment and focused investigations such as brain imaging and electrophysiological examinations can rule out these other conditions. CSF albuminocytological dissociation can occur although its absence does not preclude the diagnosis. In one study, CSF albuminocytological dissociation was present in 37% of FS and 25% of BBE patients in the first week, whereas CSF pleocytosis was present in 32% of BBE and in only 4% of FS patients.30
Pathophysiology
Anti-GQ1b antibodies
One of the major turning points in our understanding of FS and BBE came with the discovery of the IgG anti-GQ1b antibodies in typical FS patients by Chiba and colleagues.4 The authors closely followed this up by confirming the presence of anti-GQ1b in a further 18 of 19 typical FS patients as well as in five AO patients and five of six GBS patients with ophthalmoplegia, the latter representing FS overlapped by GBS.20 Other laboratories also demonstrated a similar association between IgG anti-GQ1b antibodies and FS; 83% were positive in 466 Japanese FS patients and all nine patients in the UK.15 ,42
IgG anti-GQ1b antibodies were detected in a BBE patient who was comatosed with acute ophthalmoplegia, ataxia and areflexia but made a complete recovery 2 months following the onset of her illness.5 This unexpected finding led the authors to confirm the anti-GQ1b antibodies in two other BBE patients. This common autoantibody profile in BBE and FS supported a common autoimmune mechanism in both conditions. As previously stated, other variants of FS and BBE such as AO, acute ataxic neuropathy, PCB and FS overlapped by GBS have been associated with the anti-GQ1b antibodies. It would be reasonable to collectively refer to these different neurological presentations as the ‘anti-GQ1b antibody syndrome’ to consolidate their common serological profile.7
GQ1b is highly expressed at the paranodes and the neuromuscular junctions of the oculomotor, trochlear and abducens nerves.21 ,43 Binding at these sites could explain the ophthalmoplegia and ptosis that are seen in this syndrome. Similarly, the glossopharyngeal and vagal nerves have also been shown to highly express GQ1b, accounting for oropharyngeal involvement in this syndrome.44
The pathophysiology of ataxia in the anti-GQ1b antibody syndrome continues to be subject to debate. In his original description, Fisher questioned the well-established impression at the time that the ataxia in FS was cerebellar in origin.3 He commented on the lack of pathological changes in the cerebellum and pointed out the lack of cerebellar speech in cases where there was clear jerky incoordination. Ataxia has also been demonstrated in GBS and often there is objective sensory deficit to account for ataxic neuropathy. However, the sensory involvement in FS is subtle. In their detailed electrophysiological studies of a patient with FS, Ropper and Shahani proposed a mechanism for ataxia that was purely peripheral in origin involving abnormalities of the joint position sense and muscle spindle proprioception.45 This was later supported by analyses of postural body sway in FS patients which supported distinctive sensory ataxia caused by the selective involvement of muscle spindle afferents.46 These muscle spindles contain specialised muscle fibres, which have motor innervations and are enriched with sensory endings. The neural components and intrafusal muscle fibres of these spindles have been labelled by monoclonal anti-GQ1b as well as anti-GD1b antibodies in humans and the staining pattern suggests that the group Ia afferents in muscle spindles strongly express GQ1b and GD1b.43 These group Ia afferents are the likely targets in FS patients which results in ataxia. Although GQ1b has been expressed in some large neurons in the human dorsal root ganglia,28 it is more likely that the muscle spindle targets are responsible for the ataxia experienced by FS patients. This would explain why FS patients recover without sequelae.20 The argument for a cerebellar origin to the ataxia seen in FS is still debatable. One study showed selective staining of the molecular layer of the human cerebellum by sera from FS patients who were anti-GQ1b antibody-positive.47 There have also been reported cerebellar changes on MRI of FS patients. However, MRI of FS patients are more frequently normal as demonstrated in 99% of 353 FS patients.15 Thus, at present, the evidence is weak for a central cause of ataxia.
Molecular mimicry
The presence of antecedent infections in FS and BBE was evident even from the original descriptions. Bickerstaff postulated a secondary effect rather than a direct infective illness as the likely cause of the neurological presentation in BBE. To establish a causal relationship between a microbial agent and FS or BBE, however, an epidemiological association needs to be demonstrated. In FS, a case-control study showed serological evidence of Campylobacter jejuni (21%) and Haemophilus influenzae (8%) infections were significantly more frequent in FS patients (n=73) compared with hospital controls.48 The rare occurrence of BBE makes a prospective case-control study challenging but in one retrospective study, serological evidence of a recent C jejuni (23%) or H influenzae (6%) infection in 34 BBE patients was greater when compared with hospital controls.15
In acute motor axonal neuropathy (AMAN), an axonal subtype of GBS, molecular mimicry between GM1 and C jejuni lipo-oligosaccharide has been established with the development of the AMAN rabbit model.49 A similar pathophysiology is also likely in the anti-GQ1b antibody syndrome. The lipo-oligosaccharides of C jejuni isolated from FS or BBE patients or of H influenzae isolated from an FS patient were demonstrated to mimic GQ1b.38 ,48 ,50 However, it is interesting that similar microorganisms, in this case C jejuni and H influenzae, result in the different clinical presentations that are observed in GBS and the anti-GQ1b antibody syndrome. Campylobacter sialyltransferase (CstII) is essential for the biosynthesis of ganglioside-like lipo-oligosaccharides as well as inducing the production of antiganglioside antibodies.51 C jejuni (CstII [Asn51]) strains produce GD1c- or GT1a-like lipo-oligosaccharide (figure 2).54 Patients who are infected with these C jejuni strains go on to produce IgG anti-GQ1b antibodies in patients with certain immunogenetic backgrounds. As previously discussed, binding of the anti-GQ1b antibodies to the oculomotor, trochlear and abducens nerves as well as muscle spindles in the limbs21 ,43 results in the ophthalmoplegia and ataxia that is observed in the anti-GQ1b antibody syndrome.
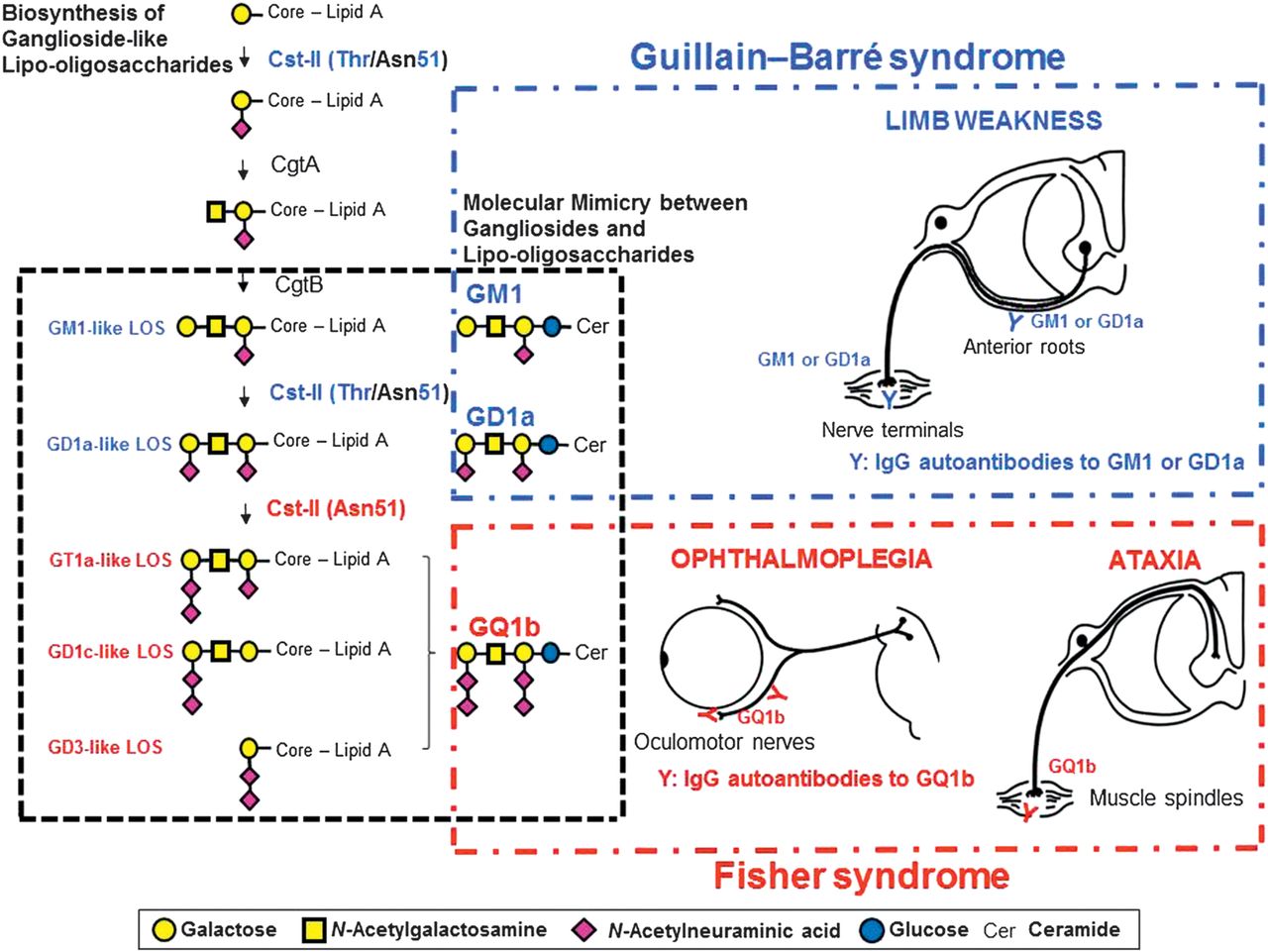
{kind=link}
{kind=link}
Molecular mechanism of the development of Fisher syndrome (FS) overlapped with Guillain–Barré syndrome (GBS) after infection by Campylobacter jejuni enteritis. The enzymes, Campylobacter sialyltransferase CstII, N-acetylgalactosaminyltransferase CgtA and galactosyltransferase CgtB, are essential for the biosynthesis of ganglioside-like lipo-oligosaccharides.52 C jejuni isolates from GBS or FS express the cstII, cgtA and cgtB genes that encode the enzymes mentioned, which are the bacterial genes responsible for the development of GBS and FS.53 The 51st amino acid of CstII determines its enzymatic activity: CstII (Thr51) produces GM1- and GD1a-like lipo-oligosaccharides, whereas Cst-II (Asn51) synthesises lipo-oligosaccharides mimicking GQ1b.52 C jejuni isolates that have cst-II (Asn51) bear lipo-oligosaccharides mimicking GQ1b, and infection by such strains may elicit the generation of immunoglobulin G (IgG) anti-GQ1b antibodies.54 The autoantibodies upon binding to GQ1b expressed in the oculomotor, trochlear and abducens nerves as well as in the muscle spindles in the limbs would cause FS.21 ,43 GT1a-like lipo-oligosaccharide is synthesised by CstII (Asn51) via GM1- and GD1a-like lipo-oligosaccharides. C jejuni strains bearing cst-II (Asn51) could induce the production of both anti-GQ1b antibodies and anti-GM1 or -GD1a antibodies. The autoantibodies bind to GM1 or GD1a expressed on the motor nerves of the limbs. Some patients with FS may develop significant limb weakness and clinical features of GBS during their course of illness. Modified from reference55 with permission.
Patients with an overlap of AMAN carry anti-GQ1b antibodies as well as anti-GM1 or -GD1a antibodies.56 In support of this, an FS isolate has been shown to carry GT1a-like lipo-oligosaccharides as well as GM1- and GD1a-like lipo-oligosaccharides as shown in the biosynthesis pathway (figure 2).54 Therefore, depending on the infective strains harboured by the susceptible patient, any clinical phenotype can be present throughout the illness and can range from the milder spectrum of incomplete FS to either FS or BBE to the more extensive cases where there is an overlap with PCB or GBS.
Demyelinating versus axonal pathology
Although GBS is known to have subtypes that are primarily demyelinating (acute inflammatory demyelinating polyneuropathy) or axonal (AMAN), there have been conflicting reports as to the nature of the underlying pathological process in FS.57 ,58 The close relationship between FS and AMAN along with its association with antiganglioside antibodies and preceding C jejuni infection would favour an axonal pathology rather than demyelination.48 ,59 Pathological studies of FS, BBE or their overlap illness with GBS have been too few and most studies have lacked sensitive tests such as teased fibre examinations or immunopathological studies to accurately differentiate demyelination from axonal pathology.2 ,14 ,27 ,60
Sequential electrodiagnostic studies are useful to distinguish among primary demyelination, axonal and reversible conduction failure of axonopathy that is occasionally seen in some forms of GBS.61 In one case series, serial neurophysiology in an FS patient showed reduced sensory amplitudes which subsequently normalised whereas in another patient with FS overlapped by GBS, initial absent sensory potentials and reduced motor amplitudes were demonstrated, in keeping with axonal neuropathy, also improving with time.62 In a separate study, serial nerve conduction studies revealed that six of 15 patients with FS and related conditions showed reversible conduction failure resembling AMAN.63 The lack of any signs of demyelination or remyelination in both studies was evident. These findings would support a primary non-demyelinating pathology in the anti-GQ1b antibody syndrome.
In the AMAN animal model, IgG antibodies have been shown to bind to nodes and paranodes and activate complement, resulting in sodium channel cluster disappearance and paranodal myelin detachment in anterior spinal nerve roots.64 Similar paranodal changes and nodal lengthening have also been demonstrated in AMAN patients.65 These pathological changes can induce conduction failure. Resolution of conduction failure and conduction block without signs of demyelination–remyelination in nerve conduction studies at recovery may explain the quick recovery in several AMAN patients. Similar to AMAN, the pathology of the anti-GQ1b antibody syndrome is likely to be due to nodal and paranodal dysfunction of the axolemma which would account for the reversible changes seen in the neurophysiology of FS patients. This would suggest that the pathophysiology of the anti-GQ1b antibody syndrome lies within the spectrum of axonal GBS.
Impaired consciousness
Patients with BBE exhibit varying degrees of altered consciousness suggesting involvement of the brainstem reticular activating system. Although one of the functions of the blood–brain barrier is to protect the brain from the deleterious effects of large molecules circulating in the bloodstream, there are several areas where the blood–brain barrier is deficient. Studies on the permeability and ultrastructure of the area postrema have demonstrated that the blood–brain barrier in this region is relatively permeable, allowing large molecules to penetrate the brainstem parenchyma at this site.66 ,67 It is possible that in BBE, the anti-GQ1b antibody reaches the brainstem via this route and attacks the brainstem reticular formation, but this has yet to be demonstrated experimentally.
Management and prognosis
The pattern of neurological characteristics seen in FS, BBE and the other subtypes of the anti-GQ1b antibody syndrome can be alarming to the clinician who is unaware of the possible underlying diagnosis. At present, the high sensitivity and specificity of the anti-GQ1b antibodies in these conditions allow the treating clinician to confirm the diagnosis. Once confirmed, the natural history of the illness is good and the majority of patients will go on to make a complete recovery even without any intervention. This was clear from the historical descriptions of these disorders. Patients who have not survived developed complications such as seizures or pneumonia rather than succumb to the actual disease.2 ,10 ,14
In a clinical review of 50 consecutive FS patients, the first symptoms to improve were ataxia (3–41 days after onset), resolving at a median of 32 days and ophthalmoplegia (between 3 and 46 days) which resolved completely at a median of 88 days.20 The majority of patients were free of ataxia and ophthalmoplegia, resuming their normal activities 6 months after neurological onset. In BBE, although most patients achieve complete remission by 6 months, symptoms of dysaesthesia, diplopia or ataxia may persist in a few patients.14
Owing to the small number of patients and its overall good prognosis, there have been no randomised clinical trials of treatment in FS or BBE.68 Both intravenous immunoglobulin and plasma exchange have been used in the treatment of both. It seems reasonable to postulate that both treatments would hasten recovery and this has been shown to be the case with intravenous immunoglobulin where there was slight hastening in recovery although the final outcome remained unchanged.69 Taking into account the cost and potential side effects of both treatments, the current recommendation would be not to treat patients unless there were clear indications to suggest possible unwarranted complications. These include patients with BBE where mortality has been reported. At the more extensive end of the anti-GQ1b spectrum, overlap with PCB or GBS would justify treatment with either intravenous immunoglobulin or plasma exchange as both have been established as efficacious in improving outcome based on randomised control trials in GBS.70 Although there have been reports of clinical improvement with steroid administration,71 its role in the anti-GQ1b antibody syndrome is less clear. Neither oral prednisolone nor intravenous methylprednisolone can significantly hasten recovery or affect the long-term outcome.72 Instead, oral corticosteroids may delay recovery. Given that the pathogenesis of anti-GQ1b antibody syndrome is similar to GBS, its use would not be recommended unless used in combination with immunoglobulin where data on the GBS population have suggested a slight hastening in recovery.73
Conclusions
It has been more than half a century since Fisher and Bickerstaff described the neurological syndromes that now bear their names. We now have a greater understanding of both disorders, in particular the underlying immunopathogenesis. To date, there is good evidence to suggest that both disorders, having similar clinical and laboratory features, are part of the same autoimmune spectrum of disorder that encompasses the peripheral and central nervous systems. Their immunological profile is also shared with other less extensive and more extensive forms. It would be justified that in pursuing our research into these conditions we refer to them by the more inclusive term ‘anti-GQ1b antibody syndrome’ to accommodate the variety of clinical presentations.
References
Footnotes
-
Funding Dr Shahrizaila receives grant support from University of Malaya (RG351/11HTM) and Dr Yuki receives support from the National Medical Research Council, Singapore (IRG 10nov086).
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.