Article Text

Abstract
Background: Distal hereditary motor neuronopathy with pyramidal features (dHMNP) is a hereditary neurodegenerative disorder characterised by the presence of upper and lower motor neuron signs. The pathophysiological mechanisms underlying these clinical findings remain elusive. Given that cortical hyperexcitability appears to underlie neurodegeneration in amyotrophic lateral sclerosis (ALS), a disorder that may clinically resemble dHMNP, the present study applied novel cortical excitability studies to further investigate the pathophysiological mechanisms in dHMNP.
Methods: Threshold tracking transcranial magnetic stimulation (TMS) studies were undertaken using a 90 mm circular coil. Peripheral nerve excitability was performed by stimulating the median nerve at the wrist, with recording made over the abductor pollicis brevis muscle. Studies were undertaken in six dHMNP and 52 ALS patients, and compared with 55 normal controls.
Results: Central motor conduction time (CMCT) was significantly prolonged in dHMNP (dHMNP 7.7 (SEM 0.7) ms; ALS 4.9 (0.3) ms; controls 5.1 (0.2) ms, p<0.01). Short interval intacortical inhibition (SICI) was significantly reduced in ALS patients (0.8 (0.8)%) when compared with dHMNP (6.4 (0.7)%, p<0.0001) and controls (8.6 (1.1)%, p<0.0001). Reduction in SICI was accompanied by significant increases in the magnetic stimulus–response curve gradient and intracortical facilitation, and reduction in cortical silent period duration in ALS, while all these parameters of cortical excitability were normal in dHMNP.
Conclusions: The present study has established a prolonged CMCT and normal cortical excitability in dHMNP, thereby providing further support for the hypothesis that cortical hyperexcitability underlies neurodegeneration in ALS.
Statistics from Altmetric.com
Hereditary motor neuronopathy comprises a heterogeneous group of disorders characterised by exclusive involvement of motor pathways, and clinically manifesting as predominantly proximal (spinal muscular atrophy) or distal (resembling Charcot–Marie–Tooth disease) neuronopathies.1 2 Distal hereditary motor neuronopathy with pyramidal features (dHMNP) is clinically characterised by the presence of upper and lower motor neuron features,3 4 5 thereby resembling amyotrophic lateral sclerosis (ALS). In ALS, it has been proposed that corticomotoneurons mediate anterior horn cell loss via an anterograde glutamate-mediated excitotoxic process.6
Cortical excitability, and thereby excitotoxicity, may be assessed using transcranial magnetic stimulation (TMS).7 Studies involving TMS have demonstrated the presence of cortical hyperexcitability in ALS.8 More recently, the threshold tracking TMS technique established that cortical hyperexcitability was an early development in sporadic ALS9 and preceded the onset of familial ALS,10 thereby suggesting that cortical hyperexcitability mediates motor neurodegeneration in ALS.
Given the clinical similarities between dHMNP and ALS, particularly the coexistence of upper and lower motor neuron signs, the present study combined novel threshold tracking studies to assess whether cortical hyperexcitability is a universal feature of degenerative motoneuron disorders, or whether it is specific to sporadic ALS.
Methods
Studies were undertaken in six patients with dHMNP from one family (four males; age range 33–65 years; mean age 46 years) with an autosomal dominant pattern of inheritance (fig 1A) and linked to a novel 7q34–q36 locus.11 All patients gave informed consent to the procedures, which were approved by the South East Sydney Area Health Service Human Research Ethics Committee.

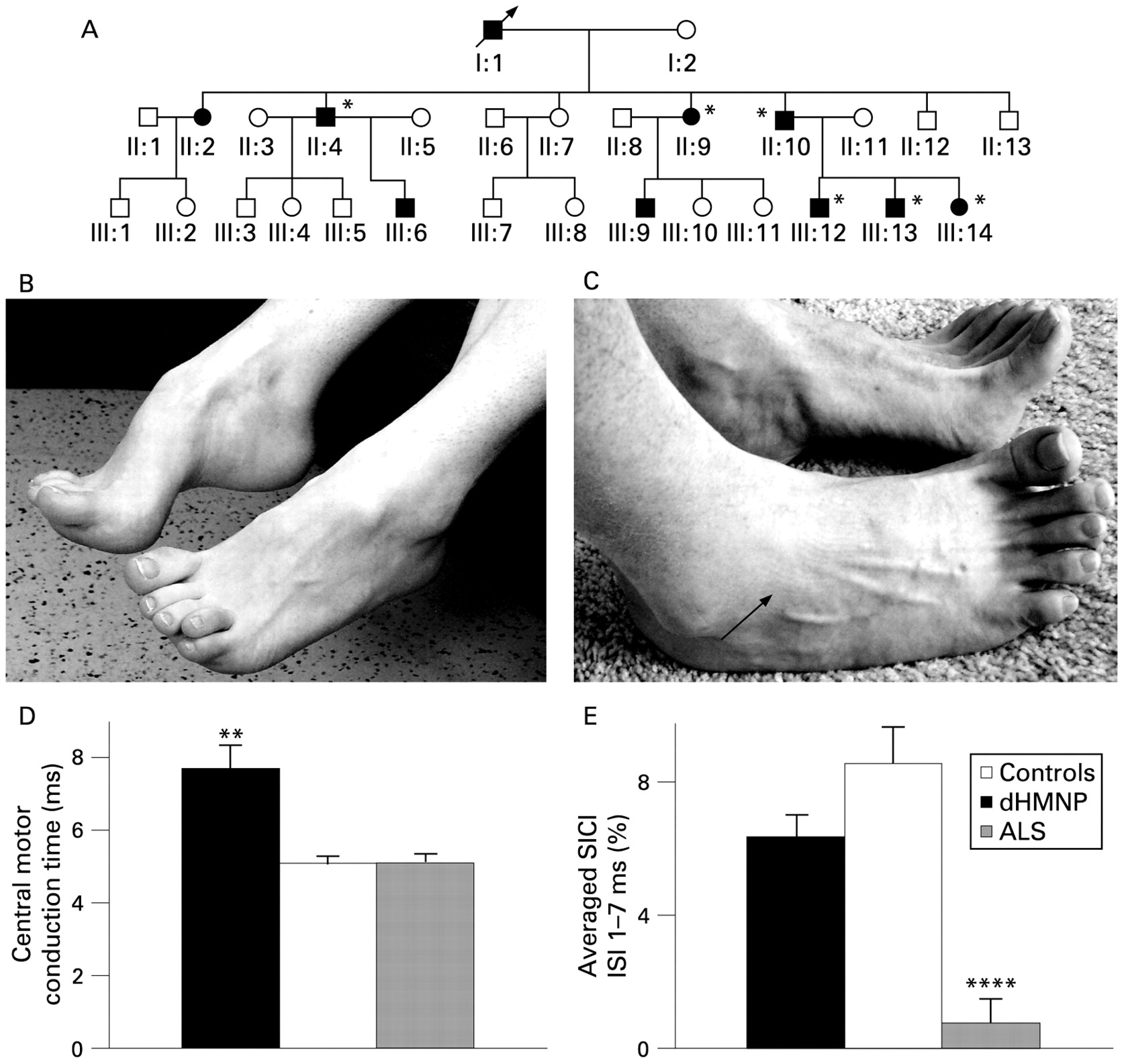{kind=link}
(A) Family pedigree illustrating an autosomal dominant pattern of inheritance in this family with distal hereditary motor neuronopathy with pyramidal tract signs. (B) Deformities of the foot, including pes cavus and hammer toes, evident in all the patients studied (*). (C) Patients exhibiting weakness of toe and ankle dorsiflexion with wasting of the extensor digitorum brevis muscle (arrowhead). (D) Central motor conduction time (ms), measured according to the F-wave method (see Methods), significantly prolonged in distal hereditary motor neuronopathy with pyramidal features (dHMNP) compared with normal controls and amyotrophic lateral sclerosis (ALS) patients. (E) Short interval intracortical inhibition (SICI), defined as the stimulus intensity required to maintain a target output of 0.2 mV (see Methods). Averaged SICI, between interstimulus intervals (ISIs) of 1 to 7 ms was significantly reduced in amyotrophic lateral sclerosis (ALS) patients when compared with patients with distal hereditary motor neuronopathy with pyramidal features (dHMNP) patients and normal controls. **p<0.01; ****p<0.0001.
Cortical excitability was measured by applying TMS to the motor cortex by means of a 90 mm circular coil. A threshold tracking paradigm was applied to record the resting motor threshold (RMT), short-interval intracortical inhibition (SICI) and intracortical facilitation (ICF) as previously described.7 In addition, single-stimulus TMS was used to record stimulus-response (SR) curves, central motor conduction time (CMCT) and cortical silent period (CSP) duration. In the same sitting, axonal excitability studies were undertaken according to a previously described protocol,12 with the median nerve stimulated at the wrist using 5 mm Ag–AgCl electrodes. The resultant peak-to-peak compound muscle action potential (CMAP) amplitude was recorded over the abductor pollicis brevis (APB), as was the neurophysiological index (NI).13
Statistical analysis
Cortical excitability in dHMNP patients was compared with control data obtained from 55 subjects (28 men; age range 23–73 years; mean 46 years) and 52 clinically probable or definite ALS patients (34 men; age range 26–71 years; mean 59 years) as defined by the revised El Escorial criteria.14 Both the control and ALS groups overlapped with a previous cohort.10 Peripheral nerve excitability parameters were compared with controls12 and ALS patients.15 All ALS patients were clinically staged using the ALS-functional rating scale—revised (ALSFRS-R),16 Medical Research Council score17 and Triggs hand function score.18 The Student t test was used to compare mean differences and analysis of variance (ANOVA) for multiple comparisons. Correlations were analysed using the Spearman rank test. A p value of <0.05 was considered statistically significant. Results are expressed as mean (SEM).
Results
Clinical features
Clinical features for dHMNP and ALS patients are summarised in table 1. An example of a foot deformity in a representative case is depicted in fig 1B,C. Brain and spinal MRI in two index cases were normal.
Clinical details for the six patients with distal hereditary motor neuronopathy with pyramidal features
Neurophysiological features
Conventional neurophysiological testing,4 11 revealed reduced median, ulnar and tibial CMAP amplitudes, and absent common peroneal nerve motor responses. Median, ulnar and sural sensory nerve action potential amplitudes were normal. EMG disclosed chronic neurogenic changes and spontaneous activity in the distal muscle groups of the upper and lower limbs.
Cortical excitability
The motor cortex was excitable in all dHMNP patients. Central motor conduction time was significantly prolonged in dHMNP patients (7.7 (0.7) ms) when compared with ALS and controls (ALS 4.9 (0.3) ms; controls 5.1 (0.2) ms, p<0.01, fig 1D).
The SR curve for TMS studies in dHMNP was significantly smaller when compared with ALS (p<0.05), but comparable when compared with controls (ANOVA, p = 0.4). There was no significant difference in RMT between the groups, which remained stable throughout the testing period.7
Short-interval intracortical inhibition
Short-interval intracortical inhibition was significantly reduced in ALS patients (0.8 (0.7)%, p<0.0001) when compared with dHMNP (6.4 (0.7)%, p<0.0001) and controls (9.0 (0.8)%, fig 1E). Peak SICI was significantly reduced in ALS patients (3.0 (1.6)%) compared with dHMNP patients (11.4 (3.4)%, p<0.05) and controls (13.7 (1.3), p<0.0001). Further, ICF was significantly reduced in dHMNP patients (−0.8 (0.5)%) when compared with ALS patients (−4.2 (0.5)%, p<0.05), but comparable with controls (−0.9 (0.4)%).
Cortical silent period
In a contracting muscle, MEP is followed by a period of electrical silence that interferes with ongoing EMG activity, known as the CSP. As the stimulus intensity increased from 60 to 150% RMT, the mean CSP duration increased from 0 to 176.7 (8.5) ms in ALS patients and was significantly shorter when compared with dHMNP patients (0–202.3 (10.2) ms, p<0.05) and controls (0–208.8 (4.8) ms, p<0.001).
Peripheral nerve excitability
The CMAP amplitude (dHMNP 7.2 (1.0) mV; ALS, 6.1 (3.6) mV; controls 10.4 (0.7) mV, p<0.05) and NI (1.4 (0.4), p<0.05; ALS, 0.8 (0.1), p<0.00001; controls, 2.5 (0.2)) were significantly reduced in dHMNP and ALS patients when compared with controls, while there was no significant difference between dHMNP and ALS patients. Further, the mean MRC from the target muscle (APB) was similar between groups (dHMNP 4.5 (0.2); ALS 4.2 (0.1)). Together, these findings indicate a comparable degree of peripheral disease burden between dHMNP and ALS patients.
The strength–duration time constant (τSD) reflects nodal persistent Na+ conductance, while rheobase is defined as the threshold current for a stimulus of infinitely long duration.19 There were no significant differences in the mean τSD and rheobase in dHMNP patients when compared with controls and ALS patients. In addition, there were no significant differences in threshold electrotonus, the recovery cycle or current–threshold relationship in dHMNP patients when compared with ALS and controls.
Correlation between cortical abnormalities, clinical and neurophysiological parameters in dHMNP
Combining measures of cortical and peripheral axonal excitability, clinical assessment and disease severity, it was evident that CMCT correlated with disease duration (R = 0.50) and the CMAP amplitude (R = −0.70). Together, these correlations suggest that prolongation of CMCT was a late feature in dHMNP.
Discussion
The present study, using a combination of novel threshold tracking techniques to explore central and peripheral processes, has established normal cortical excitability in dHMNP patients, markedly different to findings from ALS patients. Although dHMNP patients exhibited upper motor neuron dysfunction, as reflected by marked prolongation of central motor conduction time, this appeared to be a late feature of the disease, thereby suggesting that upper motor neuron dysfunction may be a secondary event. In addition, the present study has reaffirmed the presence of cortical hyperexcitability in sporadic ALS patients and supports the hypothesis that, at least with regard to progressive motor neurodegenerative disorders, cortical hyperexcitability appears specific to ALS.
Central motor conduction time
Central motor conduction time is determined by rapidly conducting large-diameter fibres.20 Although technical and physiological factors may influence CMCT,20 pathological factors such as impaired excitation of cortiomotoneurons, degeneration of large-diameter axons, excessive synaptic delay and demyelination of corticospinal axons will all result in prolongation of the CMCT.
In the present series, there was a significant prolongation of CMCT, in keeping with findings from the dHMNP phenotype due to senataxin gene mutation [ALS4].3 4 5 Although pathological studies in the ALS4 phenotype revealed demyelination of corticospinal tracts,5 the degree of CMCT prolongation in the present cohort of dHMNP patients is not diagnostic of demyelination. Specifically, in multiple sclerosis, the mean CMCT to the APB muscle has previously been reported to be 14.8 ms.21 Therefore, it may be argued that the CMCT prolongation in the present study most probably reflects degeneration of large-diameter axons. Further, the fact that CMCT correlated with CMAP amplitude and disease duration suggests that CMCT prolongation is a late feature of the disease, possibly developing as a secondary effect of the disease process.
Are the pathophysiological mechanisms underlying neurodegeneration in dHMNP similar to ALS?
Previous studies have suggested that cortical hyperexcitability underlies neurodegeneration in ALS.22 These studies have received recent support based on findings that SICI was more prominent in the early stages of disease and that cortical hyperexcitability correlated with measures of axonal loss.9 In contrast to ALS, there was no evidence of cortical hyperexcitability in dHMNP as measured by SICI, MEP amplitude, magnetic SR curve gradient and intracortical facilitation. Further, in the clinically pure lower motor neuron forms of ALS, recent studies have documented the presence of cortical hyperexcitability,23 compared with normal excitability in monomelic amyotrophy and Kennedy disease, thereby distinguishing between the different motor neuronopathies.24 25 Together, these findings suggest that despite the phenotypic similarities, cortical hyperexcitability does not underlie neurodegeneration in dHMNP but rather appears to be specific for ALS.
REFERENCES
Footnotes
SV had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding Motor Neuron Disease Research Institute of Australia (MNDRIA) and National Health and Medical Research Council of Australia (Project grant number 510233).
Competing interests None.
Ethics approval Ethics approval was provided by South East Sydney Area Health Service Human Research Ethics Committee.
Patient consent Obtained.
SV had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Provenance and Peer review Not commissioned; externally peer reviewed.